
L'alignement multiple de séquences
Contexte des écoles Wikipédia
Enfants SOS offrent un chargement complet de la sélection pour les écoles pour une utilisation sur les intranets des écoles. parrainage SOS enfant est cool!
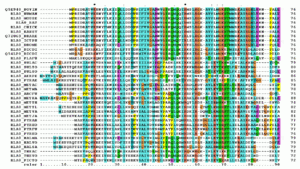
Un alignement de séquences multiples (MSA) est un alignement de séquences de trois ou plus des séquences biologiques, généralement une protéine , ADN , ou ARN. En général, l'ensemble des séquences de requêtes d'entrée sont supposés avoir une évolution relation par lequel ils partagent une lignée et descendent d'un ancêtre commun. De l'MSA, séquence résultante homologie peut être déduite et analyse phylogénétique peut être menée pour évaluer origines évolutives partagées des séquences. Représentations visuelles de l'alignement comme dans l'image à droite illustrer événements de mutation telles que des mutations ponctuelles (d'un seul acide aminé ou des modifications de nucleotides) qui apparaissent comme des caractères différents dans une colonne unique d'alignement, et l'insertion ou la deletion des mutations (ou indels) qui apparaissent comme des lacunes dans l'une ou plusieurs des séquences dans l'alignement. Alignement de séquences multiples est souvent utilisé pour évaluer séquence conservation de des domaines de protéines, tertiaire et des structures secondaires, et les acides aminés ou les nucleotides même individuels.
L'alignement multiple de séquences se rapporte également au procédé d'alignement d'un tel ensemble de la séquence. Parce que trois ou plusieurs séquences de longueur biologiquement pertinente peut être difficile et sont presque toujours du temps pour aligner à la main, de calcul des algorithmes sont utilisés pour produire et analyser les alignements. MSA exigent des méthodes plus sophistiquées que alignement par paires parce qu'ils sont plus de calculs complexes à produire. La plupart des programmes d'alignement de séquences multiples utilisent plutôt que de méthodes heuristiques optimisation globale, car l'identification de l'alignement optimal entre plus de quelques séquences de longueur modérée est prohibitivement coûteux en calcul.
La programmation dynamique et la complexité de calcul
La méthode la plus directe pour produire un MSA utilise le technique de programmation dynamique pour identifier la solution globalement optimale d'alignement. Pour les protéines, ce procédé implique généralement deux ensembles de paramètres: un pénalité de gap et un matrice de substitution attribuant des notes ou des probabilités pour l'alignement de chaque paire possible des acides aminés sur la base de la similitude des propriétés chimiques des acides aminés et la probabilité d'évolution de la mutation. Pour des séquences nucléotidiques une matrice de substitution peut être utilisé, mais comme il n'y a que quatre caractères standard possibles par séquence et les nucléotides individuels ne diffèrent généralement pas beaucoup en remplacement probabilité, les paramètres de séquences d'ADN et d'ARN se composent généralement d'une pénalité de brèche, positif score pour caractère correspond, et un score négatif pour inadéquation.
Pour n séquences individuelles, la méthode nécessite la construction de l'équivalent de dimension n de la matrice formée dans la programmation dynamique paires standard. L'espace de recherche augmente ainsi de façon exponentielle avec l'augmentation de n et est également fortement dépendant de la longueur de la séquence. Pour trouver l'optimum global pour n séquences de cette façon a été montré pour être un Problème NP-complet. Méthodes pour réduire l'espace de recherche en effectuant d'abord la programmation dynamique par paires sur chaque paire de séquences dans le jeu de requête et la recherche que l'espace de solution à proximité de ces résultats (trouver efficacement l'intersection entre les chemins locaux entourant immédiatement chaque solution optimale par paire) rendre la technique de programmation dynamique plus efficace. Le soi-disant "somme de paires" méthode a été implémentée dans le logiciel MSA, mais il est encore peu pratique pour de nombreuses applications qui nécessitent la MSA alignement simultané de plusieurs dizaines, voire quelques centaines de séquences. Méthodes de programmation dynamique sont maintenant utilisés seulement quand un alignement très haute qualité d'un petit nombre de séquences est nécessaire, et en tant que étalonnage standard dans l'évaluation de nouvelles ou raffinés techniques heuristiques.
La construction de l'alignement progressif
Un procédé d'exécution d'une recherche heuristique d'alignement progressif est la technique (aussi connu comme le procédé hiérarchique ou arborescente) qui se accumule un MSA finale en effectuant d'abord une série d'alignements par paires sur des séquences successivement moins étroitement apparentées. Ces méthodes commencent en alignant les deux séquences les plus étroitement liées d'abord, puis en alignant successivement la séquence suivante le plus étroitement lié à la requête mis à l'alignement produit dans l'étape précédente. La paire "plus liée" initial est déterminé par un système efficace méthode de classification tels que neighbor-joining basée sur une simple recherche heuristique de la requête sertie d'un outil comme FASTA. Techniques progressives donc construire automatiquement un arbre phylogénétique ainsi qu'un alignement.
Une limitation majeure des méthodes progressives est leur forte dépendance sur l'attribution initiale de parenté et sur la qualité de l'alignement initial. Les méthodes sont donc sensible aussi bien pour la distribution de séquences dans l'ensemble de la requête; la performance se améliore lorsque la parenté entre les séquences de requête est un gradient relativement lisse plutôt que lointainement grappes séparées. Les performances se dégradent aussi considérablement lorsque toutes les séquences dans le jeu sont plutôt apparenté de loin, parce inexactitudes dans l'alignement initial sont alors plus susceptibles. La plupart des méthodes progressistes modernes modifient leur fonction de score avec une fonction de pondération secondaire qui attribue des facteurs d'échelle à des membres individuels de la requête définir de façon non linéaire en fonction de leur distance phylogénétique de leurs voisins les plus proches. Choix judicieux de pondération peut aider à évaluer la parenté et atténuer les effets de relativement pauvres alignements initiaux au début de la progression.
Méthodes d'alignement progressif sont suffisamment efficaces pour mettre en œuvre sur une grande échelle pour de nombreuses séquences et sont souvent exécutés sur les serveurs Web publiquement accessibles afin que les utilisateurs ne ont pas besoin d'installer localement les applications d'intérêt. Une méthode très populaire de l'alignement progressif est le Clustal famille, en particulier la variante pondérée ClustalW dont l'accès est fourni par un grand nombre de portails Web, y compris GenomeNet, EBI, et EMBnet. Différents portails ou mises en oeuvre peuvent varier dans l'interface utilisateur et de faire différents paramètres accessibles à l'utilisateur. Clustal est largement utilisé pour la construction de l'arbre phylogénétique et comme entrée pour la structure des protéines prévision par modélisation d'homologie.
Une autre méthode de l'alignement progressif commun appelé T-Coffee est plus lente que Clustal et ses dérivés, mais produit généralement alignements plus précis pour jeux de séquences apparentées de loin. T-Coffee calcule alignements par paires en combinant l'alignement direct de la paire avec alignements indirects qui aligne chaque séquence de la paire à une troisième séquence. Il utilise la sortie de Clustal ainsi qu'un autre programme d'alignement local LALIGN qui distingue plusieurs régions de l'alignement local entre deux séquences. L'alignement obtenu et arbre phylogénétique sont utilisées comme guide pour la production de nouveaux et plus précis des facteurs de pondération.
Comme les méthodes progressistes sont heuristiques qui ne sont pas garantis à converger vers un optimum global, la qualité de l'alignement peut être difficile à évaluer et leur véritable signification biologique peut être obscure. Une méthode semi-progressive très récente qui améliore la qualité de l'alignement et ne pas utiliser une heuristique perte tout en fonctionnant en polynomiale a été mis en œuvre dans le programme PSAlign.
Méthodes itératives
Un ensemble de méthodes pour produire MSA tout en réduisant les erreurs inhérentes aux méthodes progressistes sont classés comme «itératif» parce qu'ils fonctionnent de manière similaire aux méthodes progressistes mais réaligner plusieurs reprises les séquences initiales ainsi que l'ajout de nouvelles séquences à la MSA croissante. L'une des raisons méthodes progressives sont si fortement dépendante d'un alignement initial de haute qualité est le fait que ces alignements sont toujours incorporés dans le résultat final - qui est, une fois qu'une séquence a été alignée dans la MSA, l'alignement ne est pas examinée plus avant. Cette approximation améliore le rendement au détriment de la précision. En revanche, les méthodes itératives peuvent revenir à alignements par paires précédemment calculés ou sous-MSA incorporant des sous-ensembles de la séquence de requête comme un moyen d'optimiser un général fonction objectif comme trouver un score d'alignement de haute qualité.
Une variété de subtilement différentes méthodes d'itération ont été mis en œuvre et mis à disposition dans les logiciels; commentaires et des comparaisons ont été utiles mais généralement se abstenir de choisir une technique «meilleur». Le logiciel PRRN / PRRP utilise un algorithme hill-climbing d'optimiser son score d'alignement MSA et corrige de manière itérative les deux poids d'alignement et les régions localement divergentes ou "Gappy» de la MSA croissante. PRRP fonctionne mieux lorsqu'il affiner un alignement précédemment construit par une méthode plus rapide.
Un autre programme itérative, dialign, adopte une approche inhabituelle de se concentrer étroitement sur des alignements locaux entre les sous-segments ou séquence motifs sans introduire une pénalité de brèche. L'alignement des motifs individuels est alors obtenue avec une représentation matricielle similaire à un terrain à matrice de points dans un alignement par paires. Un autre procédé qui utilise des alignements locaux rapides que les points d'ancrage ou "germes" pour une procédure globale d'alignement plus lent est réalisé sous la Suite CHAOS / dialign.
Une troisième méthode basée sur l'itération populaire appelé MUSCLE (alignement de séquences multiples par log-attente) améliore sur les méthodes progressives avec une mesure de distance plus précis pour évaluer le degré de parenté des deux séquences. La mesure de distance est mis à jour entre les étapes d'itération (bien que, dans sa forme originale, MUSCLE contenait seulement 2-3 itérations selon que le raffinement a été activé).
Modèles de Markov cachés
Modèles de Markov cachés sont des modèles probabilistes qui peuvent affecter les probabilités de toutes les combinaisons possibles de lacunes, allumettes, et l'inadéquation de déterminer la MSA le plus probable ou fixés des CES possibles. HMM peuvent produire une seule sortie plus prolifique, mais peuvent aussi générer une famille de alignements possibles qui peuvent ensuite être évalués pour signification biologique. Parce que HMM sont probabiliste, ils ne produisent pas la même solution à chaque fois qu'ils sont exécutés sur le même ensemble de données; Ainsi, ils ne peuvent pas être garantis à converger vers un alignement optimal. HMM peuvent produire des alignements à la fois globales et locales. Bien que les méthodes à base de HMM ont été développés relativement récemment, ils offrent des améliorations significatives dans la vitesse de calcul, en particulier pour les séquences qui contiennent des régions qui se chevauchent.
Typique HMM-méthodes de travail en représentant un MSA comme une forme de graphe acyclique orienté connu comme un graphe partiel d'ordre, qui consiste en une série de noeuds représentant des entrées possibles dans les colonnes d'un MSA. Dans cette représentation une colonne qui est absolument conservées (ce est que toutes les séquences dans le MSA part un caractère particulier à une position particulière) est codée comme un noeud unique avec autant de connexions sortantes qu'il ya de caractères possibles dans la colonne suivante de l'alignement. Dans les termes d'un modèle de Markov caché typique, les états observés sont les colonnes d'alignement individuels et les États «cachés» représentent la séquence ancestrale présumée à partir de laquelle les séquences dans le jeu de requête sont émis l'hypothèse d'avoir descendu. Une variante de recherche efficace du procédé de programmation dynamique, connu sous le nom Algorithme de Viterbi, est généralement utilisé pour aligner successivement le MSA croissante à l'autre dans la séquence requête fixé pour produire une nouvelle MSA. Cela est différent de méthodes d'alignement progressistes parce que le alignement de séquences antérieures est mis à jour à chaque nouvel ajout de séquence. Cependant, comme les méthodes progressives, cette technique peut être influencée par l'ordre dans lequel les séquences dans le groupe de recherche sont intégrés dans l'alignement, en particulier lorsque les séquences sont de parenté éloignée.
Plusieurs logiciels sont disponibles dans lequel les variantes des méthodes basées HMM-ont été mises en œuvre et qui sont connus pour leur évolutivité et l'efficacité, bien que correctement en utilisant une méthode de HMM est plus complexe que l'aide de méthodes progressistes les plus courantes. Le plus simple est POA (Alignement partiel-Order); un procédé similaire, mais plus généralisée est mis en oeuvre dans l'emballage SAM (alignement de séquences et de modélisation). SAM a été utilisé comme source d'alignements pour la prédiction de la structure des protéines à participer à la CASP expérience de prédiction de la structure et de développer une base de données des protéines prédites dans les levures espèces S. cerevisiae. méthodes de HMM peuvent également être utilisés pour la recherche de base de données avec HMMER.
Les algorithmes génétiques et le recuit simulé
Les techniques standard d'optimisation en informatique - qui tous deux ont été inspirés par, mais ne reproduisent pas directement, les processus physiques - ont également été utilisés dans le but de produire plus efficacement MSA qualité. Une telle technique, algorithmes génétiques, a été utilisé pour la production MSA dans une tentative pour simuler largement le processus évolutif hypothèse qui a donné lieu à la divergence dans l'ensemble de la requête. Le procédé fonctionne en brisant une série de MSA en fragments possibles et en réarrangeant les fragments de façon répétée à l'introduction de lacunes dans des positions variables. Un général fonction objectif est optimisée lors de la simulation, le plus généralement la «somme de paires" fonction de la maximisation introduit dans les méthodes de base MSA programmation dynamiques. Une technique pour les séquences de protéines a été mis en œuvre dans la saga de logiciel (alignement de séquences par l'algorithme génétique) et son équivalent dans l'ARN est appelé RAGA.
La technique de recuit simulé, par lequel un produit MSA existant par un autre procédé est affinée par une série de réarrangements visant à trouver des régions plus optimale de l'espace d'alignement que celui de l'alignement d'entrée occupe déjà. Comme la méthode des algorithmes génétiques, recuit simulé maximise une fonction objective comme la fonction de somme de paires. Recuit simulé utilise un «facteur de température" métaphorique qui détermine la vitesse à laquelle se déroulent les réarrangements et la probabilité de chaque réarrangement; typiques des périodes alterne d'utilisation du taux de réarrangement élevés relativement faible probabilité (d'explorer les régions les plus éloignées de l'espace d'alignement) avec des périodes de baisse des taux et probabilités plus élevées d'explorer plus à fond minima locaux près des régions nouvellement "colonisés". Cette approche a été mise en œuvre dans le programme Msasa (Alignement multiple de séquences par recuit simulé).
Motif conclusion

Motif constatation, également connu sous le nom d'analyse de profil, est un procédé de localisation séquence motifs de MSA mondiaux qui est à la fois un moyen de produire une meilleure MSA et un moyen de produire une matrice de notation pour une utilisation dans la recherche d'autres séquences pour des motifs similaires. Une variété de procédés pour isoler les motifs ont été développés, mais tous sont basés sur l'identification de motifs court hautement conservées au sein de l'alignement plus grande et la construction d'une matrice semblable à une matrice de substitution qui reflète la composition d'acides aminés ou de nucleotides de chaque position dans le motif putatif . L'alignement peut ensuite être affinée à l'aide de ces matrices. Dans l'analyse de profil standard, la matrice comporte des entrées pour chaque caractère possible, ainsi que des entrées pour les lacunes. Alternativement, les algorithmes de recherche de modèle statistiques peuvent identifier des motifs comme un signe précurseur d'une MSA plutôt que comme une dérivation. Dans de nombreux cas lorsque l'ensemble de la requête contient seulement un petit nombre de séquences ou de séquences contient seulement très liés, pseudocounts sont ajoutés à normaliser la distribution reflétée dans la matrice de notation. En particulier, cette corrige les entrées zéro probabilité dans la matrice à des valeurs qui sont petites mais non nulle.
Blocs analyse est une méthode de constatation motif qui limite motifs dans les régions sans brèches dans l'alignement. Les blocs peuvent être générés à partir d'un MSA ou ils peuvent être extraits à partir des séquences non alignées au moyen d'un ensemble de motifs communs précalculée précédemment générée à partir de familles de gènes connus. Bloquer le ballon se appuie généralement sur l'espacement des caractères à haute fréquence, plutôt que sur le calcul d'une matrice de substitution explicite. Le serveur BLOCS fournit une méthode interactive pour localiser ces motifs dans les séquences non alignées.
Statistique pattern-matching a été mis en œuvre en utilisant à la fois le algorithme espérance-maximisation et la Échantillonneur de Gibbs. Un des outils les plus courants de motifs d'enquête, appelées MEME, utilise maximisation des attentes et des méthodes de Markov cachés pour générer des motifs qui sont ensuite utilisés comme outils de recherche par son MAST de compagnon dans la suite combinée MEME / MAST.