
Réparation de l'ADN
Saviez-vous ...
SOS Enfants, qui se déroule près de 200 sos écoles dans le monde en développement, a organisé cette sélection. Parrainer un enfant de faire une réelle différence.
Réparation de l'ADN est un ensemble de processus par lequel une cellule détecte et corrige les dommages aux ADN qui codent pour des molécules sa génome. Dans les cellules humaines, à la fois normale activités métaboliques et les facteurs environnementaux tels que les UV et la lumière rayonnement peut causer des dommages à l'ADN, résultant en autant que 1 millions de particuliers lésions moléculaires par cellule par jour. Beaucoup de ces lésions causer des dommages structurels à la molécule d'ADN et peuvent altérer ou éliminer la capacité de la cellule à transcrire la gène qui code pour l'ADN concerné. Autres lésions induisent potentiellement dangereux des mutations dans le génome de la cellule, qui affectent la survie des cellules filles après qu'il subit la mitose. En conséquence, le processus de réparation de l'ADN est constamment actif en tant qu'elle répond à endommager la structure de l'ADN. Lorsque les processus normaux de réparation échouent, et quand cellulaire apoptose ne se produit pas, irréparables dommages à l'ADN peut se produire, y compris des cassures double brin et réticulations d'ADN (de réticule interbrins ou CIST).
Le taux de réparation de l'ADN dépend de nombreux facteurs, dont le type cellulaire, l'âge de la cellule et le milieu extracellulaire. Une cellule qui a accumulé une grande quantité de dommages à l'ADN, ou celui qui ne est plus efficace répare les dommages encourus à son ADN, peut entrer dans l'un des trois états possibles:
- un état irréversible de dormance, connu sous le nom sénescence
- suicide cellulaire, également connu sous le nom ou l'apoptose la mort cellulaire programmée
- la division cellulaire non régulée, ce qui peut conduire à la formation d'une que la tumeur est cancéreuse
La capacité de réparation de l'ADN d'une cellule est essentiel pour l'intégrité de son génome, et donc à son fonctionnement normal et celle de l'organisme. De nombreux gènes qui ont été initialement présentées à influencer la durée de vie ont avéré être impliqué dans l'ADN réparation des dommages et de la protection. Défaut de corriger lésions moléculaires dans les cellules qui forment gamètes peuvent introduire des mutations dans le génome de la descendance et influencer ainsi le taux de l'évolution .
Dommages à l'ADN
lésions de l'ADN, en raison de facteurs environnementaux et normale processus métaboliques intérieur de la cellule, se produit à une vitesse de 1000 à 1000000 lésions moléculaires par cellule par jour. Bien que cela ne constitue 0.000165% d'environ 6 milliards de bases du génome humain (3 milliards de paires de bases), des lésions non réparées dans les gènes critiques (tels que suppresseur de tumeur des gènes) peut nuire à la capacité d'une cellule à remplir sa fonction et sensiblement augmenter la probabilité de la formation de tumeurs.
La grande majorité des lésions de l'ADN affecte la structure primaire de la double hélice; autrement dit, les bases elles-mêmes sont modifiées chimiquement. Ces modifications peuvent à leur tour perturber structure hélicoïdale régulière des molécules en introduisant des liaisons chimiques non indigènes ou des adduits volumineux qui ne entrent pas dans la double hélice standard. Contrairement aux protéines et ARN, l'ADN n'a pas habituellement structure tertiaire et donc dégâts ou des perturbations ne se produisent pas à ce niveau. L'ADN est, toutefois, surenroulés protéines et enroulés autour de «packaging» appelés histones (chez les eucaryotes), et les deux superstructures sont vulnérables aux effets des dommages de l'ADN.
Types de dommages
Il ya cinq principaux types de dommages à l'ADN dus à des processus cellulaires endogènes:
- oxydation des bases [par exemple, 8-oxo-7,8-dihydroguanine (8-oxoG)] et la génération d'interruptions brins d'ADN provenant d'espèces réactives de l'oxygène,
- alkylation de bases (habituellement la méthylation), tels que la formation de la 7-méthylguanine, la 1-méthyladénine, 6-O-méthylguanine
- hydrolyse de bases, telles que désamination, dépurination et dépyrimidination.
- "La formation d'adduits volumineux" (ce est à dire, le benzo [a] pyrène diol époxyde-dG addition, aristolactam addition I-dA)
- inadéquation des bases, en raison d'erreurs dans réplication de l'ADN, dans lequel la base de l'ADN mal est cousue en place dans un brin d'ADN nouvellement formé, ou une base de l'ADN est ignorée ou insérée par erreur.
Les dommages causés par des agents exogènes prend de nombreuses formes. Certains exemples sont les suivants:
- UV-B provoque réticulation entre cytosine adjacente et bases thymine création dimères de pyrimidine. Cela se appelle dommages à l'ADN directe.
- Lumière UV-A crée des radicaux plupart gratuites. Les dommages causés par les radicaux libres est appelé dommages à l'ADN indirecte.
- Le rayonnement ionisant tel que celui créé par la désintégration radioactive ou les rayons cosmiques provoquent des ruptures dans les brins d'ADN. Rayonnements ionisants à faible niveau peut provoquer des lésions de l'ADN irréparable (conduisant à replicational et les erreurs de transcription nécessaires à la néoplasie ou peut déclencher des interactions virales) conduisant à une pré-maturité vieillissement et le cancer.
- Perturbation thermique à température élevée augmente le taux de dépurination (perte de bases de purine de l'épine dorsale de l'ADN) et lésions simple-brin. Par exemple, la dépurination hydrolytique est vu dans le bactéries thermophiles, qui poussent en sources chaudes de 40-80 ° C. Le taux de dépurination (300 résidus de purine par génome par génération) est trop élevée dans ces espèces pour être réparé par les machines de réparation normale, d'où la possibilité d'un réponse adaptative ne peut pas être exclue.
- Produits chimiques industriels tels que le chlorure de vinyle et de peroxyde d'hydrogène , et les produits chimiques environnementaux, tels que hydrocarbures aromatiques polycycliques présents dans la fumée, la suie et le goudron de créer une grande diversité de l'ADN adducts- ethenobases, bases oxydées, phosphotriesters alkylés et Réticulation de l'ADN ne en nommer que quelques-uns.
Les rayons UV, alkylation / méthylation, les dommages des rayons X et des dommages oxydatifs sont des exemples de dommages induits. Dommages spontanée peut inclure la perte d'une base, désamination, plissement de la bague de sucre et changement tautomère.
Nucléaire contre les dommages de l'ADN mitochondrial
Dans les cellules humaines, et les eucaryotes cellules en général, l'ADN se trouve dans deux des emplacements cellulaires - à l'intérieur du noyau et à l'intérieur des mitochondries . ADN nucléaire (ADN natif) existe en tant que chromatine au cours des étapes non réplicatifs du cycle cellulaire et est condensé dans les structures d'agrégats appelés chromosomes pendant la division cellulaire. Dans les deux état de l'ADN est très compacté et enroulé autour de protéines en forme de perles appelés histones. Chaque fois qu'une cellule a besoin d'exprimer l'information génétique codée dans son ADNn la région chromosomique requis est démêlé, les gènes qui s'y trouvent sont exprimés, puis la région est condensé à sa conformation de repos. ADN mitochondrial (ADNmt) est situé à l'intérieur des mitochondries organelles, existent en plusieurs exemplaires, et est également étroitement associés à un certain nombre de protéines pour former un complexe connu sous le nucléoïde. A l'intérieur des mitochondries, espèces réactives de l'oxygène (ROS), ou radicaux libres, des sous-produits de la production constante de l'adénosine triphosphate (ATP) par l'intermédiaire phosphorylation oxydative, créer un environnement hautement oxydatif qui est connue pour endommager l'ADN mitochondrial. Une enzyme essentielle dans la lutte contre la toxicité de ces espèces est la superoxyde dismutase, qui est présent à la fois dans les mitochondries et cytoplasme de cellules eucaryotes.
Sénescence et l'apoptose
La sénescence, un état irréversible dans lequel la cellule ne est plus divise, est une réponse protectrice pour le raccourcissement de la extrémités des chromosomes. Les télomères sont de longues régions répétitives ADN non codant ce plafond chromosomes et subissent une dégradation partielle chaque fois qu'une cellule subit division (voir Limite de Hayflick). En revanche, quiescence est un état réversible de la dormance cellulaire qui ne est pas liée à des dommages du génome (voir cycle cellulaire). Sénescence dans les cellules peut servir comme une alternative fonctionnelle à l'apoptose dans les cas où la présence physique d'une cellule pour des raisons spatiales est requise par l'organisme, qui sert de "dernier recours" mécanisme pour empêcher une cellule avec l'ADN endommagé de se répliquer de façon inappropriée dans le absence de pro-croissance la signalisation cellulaire. La division cellulaire non régulée peut conduire à la formation d'une tumeur (voir cancer ), qui est potentiellement mortelle pour un organisme. Par conséquent, l'induction de la sénescence et l'apoptose est considéré comme faisant partie d'une stratégie de protection contre le cancer.
dommages à l'ADN et la mutation
Il est important de distinguer les lésions de l'ADN et de mutation, les deux principaux types d'erreurs dans l'ADN. dommages de l'ADN et la mutation sont fondamentalement différents. Les dégâts sont anomalies physiques dans l'ADN, telles que les pauses simple et double brin, Résidus 8-hydroxydésoxyguanosine, et les adduits d'hydrocarbures aromatiques polycycliques. dommages à l'ADN peuvent être reconnues par des enzymes, et par conséquent, ils peuvent être correctement réparées si des informations redondantes, telle que la séquence non endommagé dans le brin d'ADN complémentaire ou homologue dans un chromosome, est disponible pour la copie. Si une cellule conserve dommages à l'ADN, la transcription d'un gène peut être empêché, et, par conséquent, la traduction en une protéine sera également bloqué. La réplication peut également être bloqué et / ou la cellule peut mourir.
Contrairement aux dommages à l'ADN, une mutation est un changement dans la séquence de bases de l'ADN. Une mutation ne peut pas être reconnu par des enzymes une fois que le changement de base est présent dans les deux brins d'ADN et, par conséquent, une mutation ne peut être réparé. Au niveau cellulaire, les mutations peuvent causer des altérations de la fonction de la protéine et de la réglementation. Les mutations sont répliquées lorsque la cellule réplique. Dans une population de cellules, les cellules mutantes vont augmenter ou diminuer la fréquence d'après les effets de la mutation sur la capacité de la cellule de survivre et se reproduire. Bien que nettement différent les uns des autres, des dommages de l'ADN et les mutations sont liées parce dommages de l'ADN provoquent des erreurs de synthèse de l'ADN lors de la réplication ou de la réparation souvent; ces erreurs sont une source majeure de mutation.
Compte tenu de ces propriétés d'endommagement de l'ADN et de la mutation, il peut être vu que les lésions de l'ADN sont un problème particulier dans la non-séparation divisant lentement ou cellules, où les dommages non réparés auront tendance à se accumuler avec le temps. D'autre part, dans les cellules à division rapide, des dommages d'ADN non réparées qui ne tuent pas les cellules en bloquant la réplication aura tendance à provoquer des erreurs de réplication et donc mutation. La grande majorité des mutations qui ne sont pas neutres dans leurs effets sont délétères pour la survie de la cellule. Ainsi, dans une population de cellules comprenant un tissu avec la réplication des cellules, les cellules mutantes ont tendance à être perdues. Cependant, des mutations rares qui fournissent un avantage de survie ont tendance à développer par clonage au détriment des cellules voisines dans le tissu. Cet avantage de la cellule est désavantageux pour l'ensemble de l'organisme, car de telles cellules mutantes peuvent donner lieu à un cancer. Ainsi, les dommages de l'ADN dans les cellules en division fréquemment, parce qu'ils donnent lieu à des mutations, sont une cause importante de cancer. En revanche, les dommages de l'ADN en divisant rarement cellules sont probablement une cause importante du vieillissement.
Les mécanismes de réparation d'ADN
Les cellules ne peuvent pas fonctionner si les dommages de l'ADN corrompt l'intégrité et l'accessibilité des informations essentielles dans le génome (mais les cellules restent en surface fonctionnelle lorsque gènes dits "non essentiels" sont manquants ou endommagés). Selon le type de dommages infligés à la structure en double hélice de l'ADN, une variété de stratégies de réparation ont évolué pour restaurer les informations perdues. Si possible, les cellules utilisent le brin complémentaire non modifiée de l'ADN ou la sœur chromatides comme matrice pour récupérer l'information originale. Sans accès à un modèle, les cellules utilisent un mécanisme de récupération d'erreurs appelée synthèse trans comme un dernier recours.
Les dommages à l'ADN modifie la configuration spatiale de l'hélice, et de telles modifications peuvent être détectées par la cellule. Une fois les dommages est localisé, des molécules spécifiques de réparation d'ADN se lient à ou près du site de la lésion, inciter d'autres molécules de lier et former un complexe qui permet la réparation réelle avoir lieu.
Inversion directe
Les cellules sont connues pour éliminer trois types de dommages à leur ADN en inversant chimiquement. Ces mécanismes ne ont pas besoin d'un modèle, étant donné que les types de dommages qu'ils neutralisent peuvent se produire dans un seul des quatre bases. Ces mécanismes d'inversion directs sont spécifiques au type de préjudice subi et ne impliquent pas la rupture du squelette phosphodiester. La formation de les dimères de pyrimidine lors de l'irradiation avec les résultats de la lumière UV dans une liaison covalente entre anormal bases pyrimidiques adjacentes. Le processus de photoréactivation inverse directement ces dommages par l'action de l'enzyme photolyase, dont l'activation est obligatoirement dépendante de l'énergie absorbée par bleu clair / UV (300-500 nm longueur d'onde) pour promouvoir la catalyse. Un autre type de dommage, la méthylation des bases guanine, est directement inverse de la protéine transférase méthyl guanine de méthyle (MGMT), l'équivalent de bactéries que l'on appelle OGT. Ce est un processus coûteux parce que chaque molécule de MGMT peut être utilisé qu'une seule fois; à savoir la réaction est stoechiométrique plutôt que catalyseur . Une réponse généralisée à des agents de méthylation dans des bactéries est connu comme le réponse adaptative et confère un niveau de résistance aux agents lors d'une exposition soutenue par alkylation régulation positive des enzymes alkylation de réparation. Le troisième type de dommages à l'ADN par les cellules inversée est certain méthylation de la cytosine et bases adénine.
Dommages simple brin
Lorsqu'un seul des deux brins d'une double hélice a un défaut, l'autre brin peut être utilisé comme un modèle pour guider la correction du brin endommagé. Afin de réparer les dommages causés à l'une des deux paires de molécules d'ADN, il existe un certain nombre de mécanismes réparation par excision de nucléotide qui éliminent endommagé et le remplacer par un nucléotide en bon état complémentaire à celle trouvée dans le brin d'ADN endommagé.
- Base de réparation excision (BER), qui répare les dommages à une base unique causés par l'oxydation, alkylation, l'hydrolyse ou désamination. La base endommagée est retirée par un ADN glycosylase. La "dent manquante" est alors reconnu par une enzyme appelée AP endonucléase, qui coupe le Liaison phosphodiester. La partie manquante est alors resynthétisé par un ADN polymérase, et une ADN ligase effectue la dernière étape nick-étanchéité.
- Excision de nucléotides réparation (TNS), qui reconnaît, lésions de l'hélice qui faussent volumineux tels que dimères de pyrimidine et 6,4 photoproduits. Une forme spécialisée de TNS connu sous le nom réparation couplée à la transcription déploie enzymes de TNS à des gènes qui sont activement transcrit.
- Réparation des mésappariements (MMR), qui corrige les erreurs de réplication de l'ADN et recombinaison qui résultent en nucléotides mispaired (mais en bon état).
Cassures double brin
Double-brin pauses, dans lequel les deux brins de la double hélice sont sectionnés, sont particulièrement dangereux pour la cellule car ils peuvent conduire à des réarrangements génomiques. Trois mécanismes existent pour réparer les cassures double brin (CDB): fin non homologue de jonction (NHEJ), microhomology médiation extrémité de jonction (MMEJ), et la recombinaison homologue. PVN Acharya a noté que cassures double-brin et une «réticulation joindre les deux brins au même point est irréparable parce que ni brin peut ensuite servir de modèle pour la réparation. La cellule va mourir dans la prochaine mitose ou, dans certains cas rares, muter ».
Dans NHEJ, L'ADN ligase IV, une société spécialisée ADN ligase qui forme un complexe avec le cofacteur XRCC4, rejoint directement les deux extrémités. Pour guider la réparation précise, NHEJ se appuie sur de courtes séquences homologues appelés microhomologies présent sur les queues simple brin de l'ADN se termine à assembler. Si ces surplombs sont compatibles, la réparation est généralement précis. NHEJ peut également introduire des mutations lors de la réparation. Perte de nucléotides endommagés sur le site de rupture peut conduire à des suppressions, et de rejoindre des formes différentes se Termini translocations. NHEJ est particulièrement important avant que la cellule a répliqué son ADN, car il n'y a pas de modèle disponible pour la réparation par recombinaison homologue. Il ya "Backup" voies de NHEJ en plus élevés eucaryotes . Outre son rôle de gardien du génome, NHEJ est nécessaire pour rejoindre cassures double-brin en épingle à cheveux coiffés induits lors V (D) J recombinaison, le processus qui génère la diversité en Des cellules B et Récepteurs des cellules T dans le vertébré système immunitaire .
La recombinaison homologue nécessite la présence d'une séquence identique ou pratiquement identique à être utilisé comme matrice pour la réparation de la cassure. Le mécanisme responsable de ce processus de réparation enzymatique est presque identique à la machine responsable croisement chromosomique pendant la méiose. Cette voie permet un chromosome endommagé pour être réparé en utilisant une sœur chromatides (disponible dans G2 après replication de l'ADN) ou d'un chromosome homologue en tant que matrice. CDB causés par la machinerie de réplication de tenter de synthétiser sur un saut simple brin ou réparé la cause de la lésion effondrement du fourche de réplication et sont généralement réparés par recombinaison.
Topoisomérases introduire deux pauses simple et double-brin dans le cadre de l'évolution de l'état de l'ADN de superenroulement, qui est particulièrement fréquente dans les régions à proximité d'une fourche de réplication ouverte. Ces pauses ne sont pas considérés comme des dommages de l'ADN, car ils sont un intermédiaire naturel dans le mécanisme biochimique de la topoisomérase et sont immédiatement réparées par les enzymes qui les ont créés.
Une équipe de chercheurs français a bombardé Deinococcus radiodurans pour étudier le mécanisme de double brin réparation de l'ADN de rupture dans cet organisme. Au moins deux copies du génome, avec des pauses aléatoires d'ADN peuvent former des fragments d'ADN à travers recuit. Partiellement fragments se chevauchent sont ensuite utilisés pour la synthèse des des régions homologues à travers un déplacement D-boucle qui peut continuer l'extension jusqu'à ce qu'ils trouvent brins complémentaires des partenaires. Dans la dernière étape, il est croisement au moyen de RecA-dépendante la recombinaison homologue.
synthèse trans
synthèse trans (TLS) est un procédé de tolérance aux dommages à l'ADN qui permet la machinerie de replication d'ADN à répliquer dernières lésions de l'ADN, tels que dimères de thymine ou Sites AP. Il se agit de commutation sur régulière Les ADN polymerases de polymérases spécialisées translésionnelles (c.-à ADN polymerase IV ou V, la polymérase de la famille Y), souvent avec des sites actifs plus importantes qui peuvent faciliter l'insertion des bases opposées nucleotides endommagés. La commutation de la polymérase est censée être médiée par, entre autres facteurs, la modification post-traductionnelle de la replication facteur de processivité PCNA. polymérases synthèse trans ont souvent une faible fidélité (forte propension à insérer bases mauvaises) sur les modèles bon état par rapport à polymérases réguliers. Cependant, beaucoup sont extrêmement efficaces pour l'insertion des bases correctes opposés types spécifiques de dommages. Par exemple, Pol η médie dérivation sans erreur des lésions induites par l'irradiation UV , alors que Pol ι introduit des mutations dans ces sites. Pol η est connu d'ajouter la première adénine à travers la T ^ T photodimère aide Watson-Crick de base et la seconde adénine seront ajoutées dans sa conformation syn l'aide Appariement de bases de Hoogsteen. Du point de vue cellulaire, risquer l'introduction de mutations ponctuelles pendant synthèse trans peut être préférable de recourir à des mécanismes plus drastiques de réparation de l'ADN, ce qui peut provoquer des aberrations chromosomiques bruts ou mort cellulaire. En bref, le processus implique spécialisée polymérases soit en contournant ou la réparation des lésions à des endroits de réplication de l'ADN au point mort. ADN polymérase eta peut contourner lésions de l'ADN par exemple trop complexes guanine-thymine réticulation intra-brin, G [8,5-Me] T, bien que peuvent causer des mutations ciblées et semi-ciblée. La toxicité et la mutagenèse de cette lésion a été étudiée par la réplication d'un plasmide G [8,5-Me] T modifié dans Escherichia coli avec KO spécifiques de l'ADN polymérase. La viabilité était très faible dans une souche dépourvue pol II, pol IV, V et pol, les ADN polymerases trois SOS-inductibles, ce qui indique que synthèse trans est effectuée principalement par les ADN polymérases. Une plate-forme de dérivation est fournie pour ces polymérases par Antigène nucléaire de prolifération cellulaire (PCNA). Dans des circonstances normales, PCNA lié à l'ADN polymérases réplique. Sur un site de lésion, PCNA est ubiquitinée, ou modifiée, par le RAD6 / RAD18 protéines pour fournir une plate-forme pour les polymérases spécialisées de contourner la lésion et reprendre la réplication de l'ADN. Après synthèse trans, extension est nécessaire. Cette extension peut être effectuée par une polymérase réplicative si le TLS est sans erreur, comme dans le cas de Pol η, mais si TLS se traduit par un décalage, une polymérase spécialisée est nécessaire de l'étendre; Pol ζ. Pol ζ est unique en ce qu'elle peut se étendre inadéquation terminaux, alors que polymérases plus processives ne peut pas. Alors, quand une lésion est détectée, la fourche de réplication freinera, PCNA se passer d'une polymérase processive à une polymérase TLS tels que Pol ι de fixer la lésion, puis PCNA peut passer à Pol ζ d'étendre l'inadéquation et PCNA dernière passe à la polymérase de continuer la réplication processive.
Réponse globale aux dommages de l'ADN
Les cellules exposées à rayonnements ionisants, la lumière ultraviolette ou de produits chimiques sont susceptibles d'acquérir plusieurs sites de lésions de l'ADN volumineux et cassures double brin. En outre, les agents endommageant l'ADN peut endommager les autres des biomolécules telles que des protéines , des glucides , des lipides , et ARN. L'accumulation de dommages, d'être, les pauses ou des adduits de décrochage du double brin spécifiques fourches de réplication, sont parmi signaux connus de stimulation pour une réponse globale aux dommages de l'ADN. La réponse mondiale à des dommages est un acte dirigé vers propre conservation des cellules et déclenche de multiples voies de réparation macromoléculaire, lésion dérivation, la tolérance, ou apoptose. Les caractéristiques communes de la réponse mondiale sont induction de multiples gènes, l'arrêt du cycle cellulaire, et l'inhibition de la division cellulaire.
Lésion de l'ADN des points de contrôle
Après dommages à l'ADN, cycle cellulaire points de contrôle sont activés. Activation Checkpoint arrête le cycle cellulaire et donne le temps de la cellule pour réparer les dégâts avant de continuer à se diviser. lésion de l'ADN se produisent des points de contrôle à la G1 / S et G2 / M limites. Un intra- S checkpoint existe aussi. Checkpoint activation est contrôlé par deux maîtres kinases, ATM et ATR. ATM répond aux cassures double-brin et des perturbations dans la structure de la chromatine, alors que ATR répond essentiellement à l'impasse fourches de réplication. Ces kinases phosphoryler cibles en aval dans un transduction du signal en cascade, pour aboutir finalement à l'arrêt du cycle cellulaire. Une classe de médiateurs protéiques de point de contrôle comprenant BRCA1, MDC1, et 53BP1 a également été identifiée. Ces protéines semblent être requis pour transmettre le signal de point de contrôle pour l'activation de protéines en aval.
Une cible en aval importante de l'ATM et ATR est p53, comme il est nécessaire pour induire apoptose suite aux dommages de l'ADN. Le inhibiteur de la kinase cycline-dépendante p21 est induite par des mécanismes dépendant de p53 et p53-indépendante et peut arrêter le cycle cellulaire en G1 / S et G2 / M points de contrôle en désactivant cycline / des complexes de kinases cycline-dépendantes.
La réponse SOS procaryote
Le SOS réponse est les changements dans l'expression du gène dans Escherichia coli et d'autres bactéries, en réponse à des dégâts à l'ADN. Le SOS système procaryote est régi par deux protéines clés: LexA et RecA. Le LexA est un homodimère transcriptionnelle répresseur qui se lie à séquences d'opérateur communément appelés boîtes SOS. En Escherichia coli, il est connu que LexA régule la transcription de gènes, y compris environ 48 gènes recA et lexA. La réponse SOS est connu pour être très répandue dans le domaine de bactéries, mais ce est surtout l'absence dans certains phylums bactérienne, comme le Spirochètes. Les signaux cellulaires les plus courantes activant la réponse SOS sont des régions de l'ADN simple brin (ADNsb), découlant de l'impasse fourches de réplication ou cassures double-brin, qui sont traités par ADN hélicase à séparer les deux brins d'ADN. Dans l'étape d'initiation, se lie à la protéine RecA dans un ADNsb Hydrolyse de l'ATP conduit réaction création filaments RecA-ADNsb. Filaments RecA-ADNsb activer LexA automatique activité de la protease, ce qui conduit finalement à un clivage de LexA dimère et la dégradation subséquente de LexA. La perte de répresseur LexA induit la transcription des gènes SOS et permet en outre l'induction de signal, l'inhibition de la division cellulaire et une augmentation des niveaux de protéines responsables de la transformation des dommages.
Dans Escherichia coli, boîtes SOS sont 20 longues séquences nucléotidiques près de promoteurs avec la structure palindromique et un degré élevé de conservation de séquence. En d'autres classes et embranchements, la séquence de boîtes SOS varie considérablement, avec une longueur et une composition différente, mais il est toujours hautement conservé et l'un des signaux les plus forts courtes dans le génome. La haute teneur en informations des boîtes SOS permet la liaison de LexA différentiel différents promoteurs et permet pour la synchronisation de la réponse SOS. Les gènes de réparation de lésion sont induites au début de la réponse SOS. Les translésionnelles polymérases sujettes à l'erreur, par exemple, UmuCD'2 (polymerase V également appelé ADN), sont induites par la suite comme un dernier recours. Une fois les dommages de l'ADN est réparé ou contourné en utilisant polymérases ou par recombinaison, la quantité d'ADN simple brin dans les cellules est diminué, réduisant les quantités de filaments RecA diminue l'activité de clivage de LexA homodimère, qui se lie aux boîtes SOS près de promoteurs et restaurations l'expression du gène normal.
Réponses de transcription eucaryotes aux dommages de l'ADN
Eucaryotes cellules exposées à des agents endommageant l'ADN activent également les voies de défense importants en induisant plusieurs protéines impliquées dans la réparation de l'ADN, cycle cellulaire de commande de point de contrôle, le trafic de la protéine et de la dégradation. Cette génome large réponse transcriptionnelle est très complexe et étroitement réglementé, permettant ainsi réponse mondiale coordonnée aux dommages. L'exposition de levure Saccharomyces cerevisiae à l'ADN des agents endommageant résultats dans chevauchent mais les profils de transcription distincts. Similitudes avec l'environnement réponse au choc indique qu'une voie de réponse au stress mondiale générale existe au niveau de l'activation transcriptionnelle. En revanche, les différents types de cellules humaines répondent aux dommages indiquant différemment l'absence d'une réponse globale commune. L'explication probable de cette différence entre la levure et les cellules humaines peut être dans le hétérogénéité des mammifères cellules. Chez un animal de différents types de cellules sont réparties entre les différents organes qui ont évolué des sensibilités différentes à des dommages de l'ADN.
En réponse global général à des lésions de l'ADN implique l'expression de multiples gènes responsables réparation postreplication, recombinaison homologue, la réparation par excision de nucléotides, Dommages à l'ADN checkpoint, activation globale de la transcription, gènes contrôlant la dégradation des ARNm, et bien d'autres. Une grande quantité de dégâts à une cellule laisse avec une décision importante: apoptose et mourir ou survivre au coût de la vie avec un génome modifié. Une augmentation de la tolérance aux dommages peut conduire à une augmentation du taux de survie qui permettra une plus grande accumulation de mutations. Levure Rev1 et polymérase humaine η sont membres de [ADN translésionnelles de famille Y polymérases présents lors réponse globale aux dommages de l'ADN et sont responsables de mutagenèse accrue lors d'une réponse globale aux dommages de l'ADN chez les eucaryotes.
réparation de l'ADN et le vieillissement
Effets pathologiques d'une mauvaise réparation de l'ADN
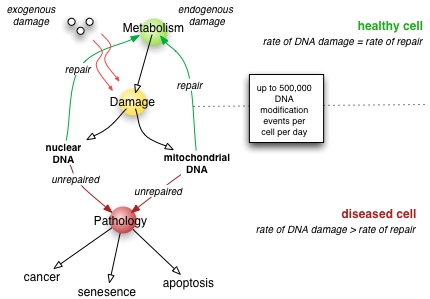
Les animaux de laboratoire avec des déficiences génétiques en réparation de l'ADN souvent montré une diminution de la durée de vie et l'incidence accrue du cancer. Par exemple, les souris déficientes dans la voie NHEJ dominante et télomères mécanismes de maintenance se lymphome et les infections plus souvent, et, par conséquent, avoir une durée de vie plus courte que les souris de type sauvage. De manière similaire, les souris déficientes en une réparation et la transcription protéine clé qui se déroule hélices d'ADN ont apparition prématurée des maladies liées au vieillissement et raccourcissement de la durée de vie. Cependant, pas tous la carence de réparation d'ADN crée exactement les effets prévus; Les souris déficientes dans la voie de TNS exposé raccourci la durée de vie sans hausse correspondante des taux de mutation.
Si le taux de dommages à l'ADN dépasse la capacité de la cellule à réparer, l'accumulation d'erreurs peut submerger la cellule et entraîner au début de la sénescence, l'apoptose, ou le cancer. Maladies associées à défectueuse réparation de l'ADN fonctionne résultat dans le vieillissement prématuré Hérité, une sensibilité accrue à des agents cancérigènes, et le risque accru de cancer en conséquence (voir ci-dessous ). D'autre part, les organismes améliorés avec les systèmes de réparation de l'ADN, tels que Deinococcus radiodurans, l'organisme le plus résistant aux radiations connue, présentent une résistance remarquable aux effets cassures double-brin induisant de la radioactivité, probablement due à une meilleure efficacité de réparation de l'ADN et surtout NHEJ.
La longévité et la restriction calorique
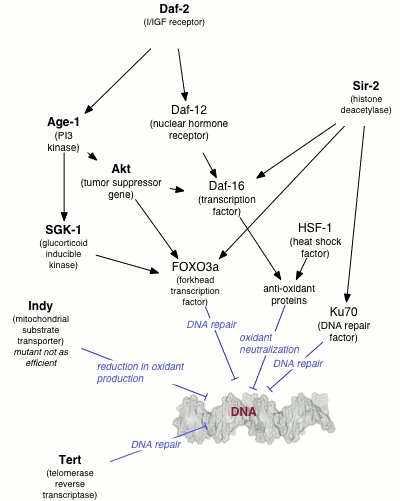
Un certain nombre de gènes individuels ont été identifiés comme ayant une influence des variations de la durée de vie au sein d'une population d'organismes. Les effets de ces gènes sont fortement dépendantes de l'environnement, en particulier, sur l'alimentation de l'organisme. La restriction calorique se traduit de manière reproductible dans la durée de vie prolongée dans une variété d'organismes, probablement via voies de détection en nutriments et une diminution taux métabolique. Les mécanismes moléculaires par lesquels ces résultats de restriction dans la durée de vie allongée sont encore floues (voir pour une discussion); Cependant, le comportement de nombreux gènes connus pour être impliqués dans la réparation de l'ADN est modifiée dans des conditions de restriction calorique.
Par exemple, l'augmentation de la dosage génique du gène SIR-2, qui réglemente l'emballage de l'ADN dans les ver nématode Caenorhabditis elegans, peut se étendre de manière significative la durée de vie. L'homologue mammifère de la SIR-2 est connue pour induire en aval facteurs de réparation d'ADN impliquées dans NHEJ, une activité qui est particulièrement encouragée dans des conditions de restriction calorique. La restriction calorique a été étroitement lié au taux de l'excision de base de réparation de l'ADN nucléaire de rongeurs, bien que les effets similaires ne ont pas été observées dans l'ADN mitochondrial.
Il est intéressant de noter que l'C. elegans gène AGE-1, un effecteur amont des voies de réparation de l'ADN, confère considérablement prolongé la durée de vie dans des conditions de libre-alimentation mais conduit à une diminution de la capacité de reproduction dans des conditions de restriction calorique. Cette observation appuie le théorie de la pléiotropie origines biologiques du vieillissement, ce qui suggère que les gènes conférant une grande avantage de survie tôt dans la vie seront sélectionnés pour même se ils portent un désavantage correspondant tard dans la vie.
Médecine et réparation de l'ADN modulation
Troubles de réparation d'ADN héréditaires
Des défauts dans le mécanisme de TNS sont responsables de plusieurs maladies génétiques, y compris:
- Xeroderma pigmentosum: hypersensibilité à la lumière du soleil / UV, entraînant une augmentation de l'incidence du cancer de la peau et le vieillissement prématuré
- Syndrome de Cockayne: hypersensibilité aux rayons UV et aux agents chimiques
- Trichothiodystrophie: la peau sensible, les cheveux et les ongles cassants
Le retard mental accompagne souvent les deux derniers troubles, ce qui suggère une plus grande vulnérabilité des neurones de développement.
Autres troubles de réparation d'ADN comprennent:
- Le syndrome de Werner: vieillissement prématuré et un retard de croissance
- La lumière du soleil l'hypersensibilité, la forte incidence du syndrome de Bloom des tumeurs malignes (en particulier les leucémies).
- Ataxie télangiectasie: la sensibilité aux rayonnements ionisants et des agents chimiques
Toutes les maladies ci-dessus sont souvent appelés «segmentaire progerias "(" vieillissement accéléré maladies ") parce que leurs victimes apparaissent personnes âgées et souffrent de maladies liées au vieillissement à un anormalement jeune âge, tout en ne manifestant tous les symptômes de la vieillesse.
D'autres maladies associées à la fonction de réparation d'ADN réduite comprennent L'anémie de Fanconi, héréditaire cancer du sein héréditaire et cancer du côlon.
réparation de l'ADN et le cancer
En raison des limites inhérentes aux mécanismes de réparation d'ADN, si les humains vivaient assez longtemps, ils seraient tous éventuellement développer un cancer. Il existe au moins 34 Hérité des mutations de gènes de réparation d'ADN humains qui augmentent le risque de cancer. Plusieurs de ces mutations provoquent réparation de l'ADN d'être moins efficace que la normale. En particulier, Héréditaire cancer polypose colorectal (HNPCC) est fortement associée à des mutations spécifiques dans le mismatch repair voie. BRCA1 et BRCA2, deux gènes célèbres dont les mutations confèrent un risque extrêmement élevé de cancer du sein sur les transporteurs, sont tous deux associés à un grand nombre de voies de réparation d'ADN, NHEJ et en particulier la recombinaison homologue.
les procédures de traitement du cancer, tels que la chimiothérapie et de radiothérapie travail écrasante de la capacité de la cellule à réparer les dommages de l'ADN, ce qui entraîne la mort cellulaire. Les cellules qui sont les plus divisent rapidement - le plus souvent des cellules cancéreuses - sont touchées préférentiellement. L'effet secondaire est que d'autres cellules non cancéreuses à division rapide, mais telles que des cellules souches de la moelle osseuse sont également affectés. Traitements du cancer modernes tentent de localiser la lésion de l'ADN à des cellules et des tissus uniquement associés au cancer, soit par des moyens physiques (concentration de l'agent thérapeutique dans la région de la tumeur) ou par des moyens biochimiques (qui exploitent une caractéristique unique de cellules cancéreuses dans l'organisme) .
réparation de l'ADN et de l'évolution
Les processus de base de réparation de l'ADN sont très conservée parmi les procaryotes et les eucaryotes et même parmi les bactériophages ( des virus qui infectent les bactéries ); Toutefois, des organismes plus complexes avec plus de génomes complexes disposent de mécanismes de réparation en conséquence plus complexes. La capacité d'un grand nombre de protéines motifs structuraux à catalyser des réactions chimiques appropriées a joué un rôle important dans l'élaboration de mécanismes de réparation au cours de l'évolution. Pour un examen extrêmement détaillée des hypothèses relatives à l'évolution de la réparation de l'ADN, voir.
Le registre fossile indique que la vie seule cellule a commencé à proliférer sur la planète à un moment donné au cours de la précambrien période, mais quand exactement reconnue par la vie moderne est apparue est claire. Les acides nucléiques sont devenus le moyen unique et universel de codage de l'information génétique, nécessitant réparation de l'ADN mécanismes qui dans leur forme de base ont été héritées par toutes les formes de vie existantes de leur ancêtre commun. L'émergence de l'atmosphère riche en oxygène de la Terre (connue comme la " catastrophe de l'oxygène ") en raison de photosynthétiques organismes, ainsi que la présence de potentiellement dommageables des radicaux libres dans la cellule en raison de la phosphorylation oxydative, a nécessité l'évolution des mécanismes de réparation d'ADN qui agissent spécifiquement pour contrer les types de dommages induits par le stress oxydatif.
Taux de changement évolutif
Dans certains cas, les dommages de l'ADN n'a pas été réparé, ou est réparé par un mécanisme d'erreurs qui se traduit par un changement de la séquence d'origine. Lorsque cela se produit, des mutations peuvent se propager dans les génomes de la progéniture de la cellule. Si un tel événement se produira dans une cellule germinale de ligne qui finira par produire un gamète, la mutation a le potentiel pour être transmis à la descendance de l'organisme. Le taux d' évolution dans une espèce particulière (ou, dans un gène particulier) est une fonction du taux de mutation. En conséquence, le taux et la précision des mécanismes de réparation d'ADN ont une influence sur le processus de changement évolutif.